
 #Notícias
#Notícias
#Notícias
13.06.2023
Riscos da aprovação acelerada
Estados Unidos autorizam tratamentos de câncer oito meses antes da Europa, mas desempenho deve ser visto com ressalvas, indica estudo
 Crédito: Bruna Martins / Estúdio Voador
Crédito: Bruna Martins / Estúdio Voador
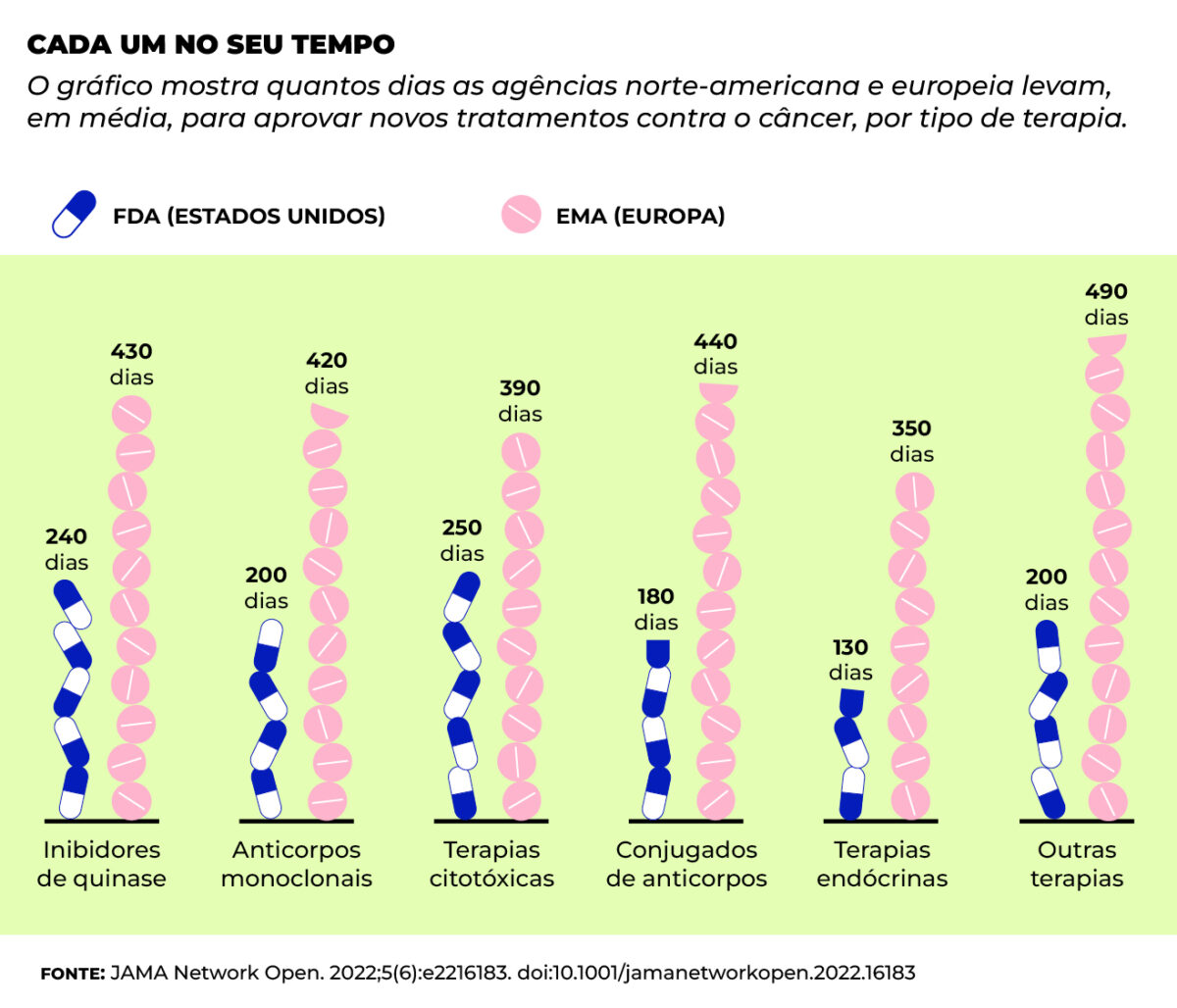
Investigação realizada por pesquisadores de Estados Unidos, Canadá e Reino Unido revelou
que, entre 2010 e 2019, considerando 89 novas terapias oncológicas aprovadas pelas agências
regulatórias dos Estados Unidos (Food and Drug Administration – FDA) e da Europa (European
Medicines Agency – EMA), a instituição norte-americana aprovou 95% dos fármacos primeiro,
enquanto a União Europeia autorizou a comercialização em média 241 dias depois.
Os resultados foram publicados no periódico JAMA Network Open. De acordo com o
autor principal do estudo, o farmacologista Mark Lythgoe, do Imperial College de Londres, no
Reino Unido, ainda hoje o “atraso” europeu permanece relativamente inalterado em relação a
uma década atrás. Oito meses de diferença é um período altamente significativo, uma vez que
uma espera tão longa pode exceder a expectativa de vida de muitas pessoas com câncer
avançado, pondera o especialista britânico.
“A aprovação mais acelerada pela FDA permitiu o acesso a muitos medicamentos inovadores
que transformaram positivamente a vida de milhares de pacientes”, disse Lythgoe ao ScienceArena. Como exemplo, ele cita o antitumoral imatinib – atualmente comercializado pela
multinacional farmacêutica Novartis como Gleevec –, que transformou o tratamento da
leucemia mieloide crônica, um tipo raro de câncer que atinge células do sangue.
“Trata-se, no entanto, de um cenário misto, com duas faces da mesma moeda”, ressalva o
pesquisador. Em comparação com a atuação da EMA, mais medicamentos oncológicos novos,
aprovados, precisaram ser posteriormente retirados de mercado pela FDA. Isso pode indicar
que períodos de revisão mais curtos nem sempre se traduzem em melhores resultados.
Lythgoe menciona outro estudo, publicado em 2019 também no JAMA, que mostrou que de
93 medicamentos contra o câncer com aprovação acelerada da FDA, apenas 19 (20%)
demonstraram melhora de sobrevida. “Velocidade e volume de aprovações só devem ser
celebrados se entregarem resultados mais significativos, fazendo com que o paciente viva
melhor e por mais tempo”, diz Lythgoe.
Ali Raza Khaki, oncologista da Escola de Medicina da Universidade Stanford, na Califórnia, e
coautor do paper, ressalta que as descobertas do grupo não podem ser diretamente
relacionadas ao nível de burocracia de nenhuma das agências reguladoras, nem às intenções
mais ou menos cuidadosas dos órgãos.
De 93 medicamentos oncológicos com aprovação acelerada da FDA, apenas 19 demonstraram melhora de sobrevida.
Contudo, os dois pesquisadores chamam a atenção para uma particularidade do processo de
análise adotado pela EMA em duas etapas. A primeira é a exigência de aprovação pelo Comitê
de Medicamentos para Uso Humano. Em seguida, é mandatória a adoção pela Comissão
Europeia, uma aprovação centralizada. Só esta segunda etapa provoca acréscimo de 62 dias
nos trâmites. Em contraste, a FDA adota vias de análise “turbinadas”, entre as quais se destaca
a chamada “breakthrough designation”, em que se reconhece liminarmente que um
determinado fármaco conquistou um patamar elevado de ação terapêutica.
Mas se, por um lado a EMA retirou menos medicamentos do mercado, por outro aprovou
menos medicamentos, em comparação com a FDA, antes da publicação científica de
embasamento (landmark publication). “Consideramos isso importante, porque permite que a
comunidade médica tenha em mãos os dados sobre a população que provavelmente se
beneficiará, além de informações completas e claras sobre benefícios, riscos da terapia e
gerenciamento adequado da toxicidade”, explicou Khaki ao ScienceArena.
Incertezas sobre benefício
Limitações da aprovação acelerada de medicamentos também foram ressaltadas por Kristina
Jenei, pesquisadora da Faculdade de População e Saúde Pública da Universidade da Colúmbia
Britânica, em Vancouver, no Canadá. Em comentário publicado no JAMA, Jenei argumentou
que a aprovação mais rápida deixa mais incertezas sobre os reais benefícios do tratamento,
expondo pacientes a riscos adicionais, como toxicidade indevida de medicamentos.
Isso é agravado, diz Jenei, pela lentidão de estudos de acompanhamento e confirmação de
benefício clínico pós-comercialização. A pesquisadora também alerta para sobre um desafio
subestimado, em suas palavras. “O aumento da velocidade de aprovação nos EUA combinado
a um follow up inconsistente após a liberação do medicamento contribui para diminuir os
padrões globais de análise de fármacos”, escreveu.
Finalmente, lembra Jenei, os Estados Unidos têm o maior mercado farmacêutico do mundo e,
portanto, são prioridade para os fabricantes em suas estratégias de definição de preços. De
fato, 72% das empresas do setor apresentaram solicitações de aprovação de fármacos ao FDA
antes de submetê-las à EMA.
“Essa abordagem pode demonstrar uma tática corporativa para lançar medicamentos em
países dispostos a pagar preços mais altos, o que, por sua vez, aumenta os preços
globalmente. Outros países devem negociar preços de medicamentos que foram projetados
para o mercado americano”, afirmou Jenei.
*
É permitida a republicação das reportagens e artigos em meios digitais de acordo com a licença Creative Commons CC-BY-NC-ND.
O texto não deve ser editado e a autoria deve ser atribuída, incluindo a fonte (Science Arena).



